I’m trying to customize my MultiQC report. In particular, I would like to personalize the content of the General Stats table by including specific columns and removing others. We have already made several attempts without success, including using the custom_data option in the multiqc_config.yaml.
I’m attaching my multiqc_config.yaml here in the hope of getting some suggestions from you.
Before I go into the specifics of your question, can I back up a bit and ask what it is that you’re trying to do? Your config file is passing in custom data (ie. not supported by MultiQC), but from supported tools (eg. FastQC and STAR).
If you want to hide columns in general stats, you can just toggle them off using table_columns_visible (see docs).
Or if you generally want to control the columns in general stats (eg. order, titles etc) then you want general_stats_columns (docs) (can set hidden: true in here to hide columns too).
From what you’ve written, I guess that these should achieve what you want and that you don’t need custom_data at all. Note that the top-level star in your config is not a valid key so is just being ignored, you can remove that too.
I hope this helps! Let me know if I missed something.
Thanks for your kind reply.
To answer your question: I built a pipeline that takes FASTQ.GZ files, performs trimming, and runs FastQC on both the raw and trimmed data. Then, the trimmed_reads are aligned with STAR on hg38 genome. After that, the workflow branches into two paths: we performe HTSeq count both on the aligned reads and on the deduped_reads (UMI_TOOLS). The goal is to compare the number of detected genes with and without deduplication.
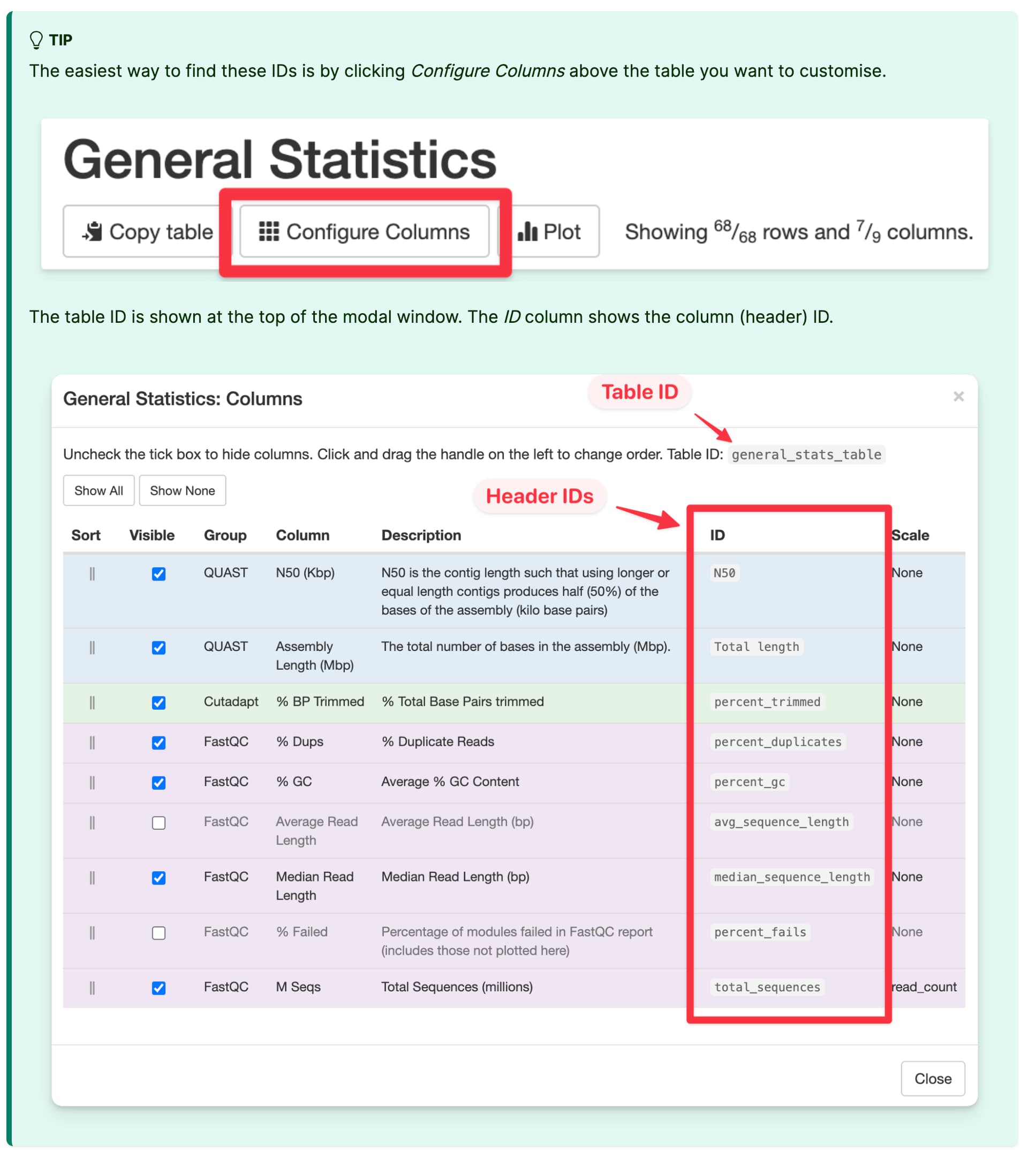
I tried to follow your advice, using table_columns_visible and general_stats_columns but I still have the same issues. Actually, we’re not confident about the columns’ IDs that we are specifying in table_column_visible. However, we don’t know how to retrieve this information.
Moreover, we’d like to add info about detected genes present in 2 files generated with our pipeline. The files’ name are: HTSEQCOUNT_STAR_detectedgenes.tsv and HTSEQCOUNT_DEDUP_detectedgenes.tsv; both contain column name Detected_Genes.
This is best done using Custom Content - set plot_type: "generalstats" to get numbers into the general statistics table. You’re nearly there in your config file, there are just a couple of errors.
These need to be nested under a sp key, short for search pattern (see docs). Currently they are being ignored, which means that the custom_content block can’t find any files to work with.
Other stuff: it doesn’t really make sense to specify both fn_clean_exts and extra_fn_clean_exts - the former overwrites all the defaults, the latter appends to them. You only need to use one type.