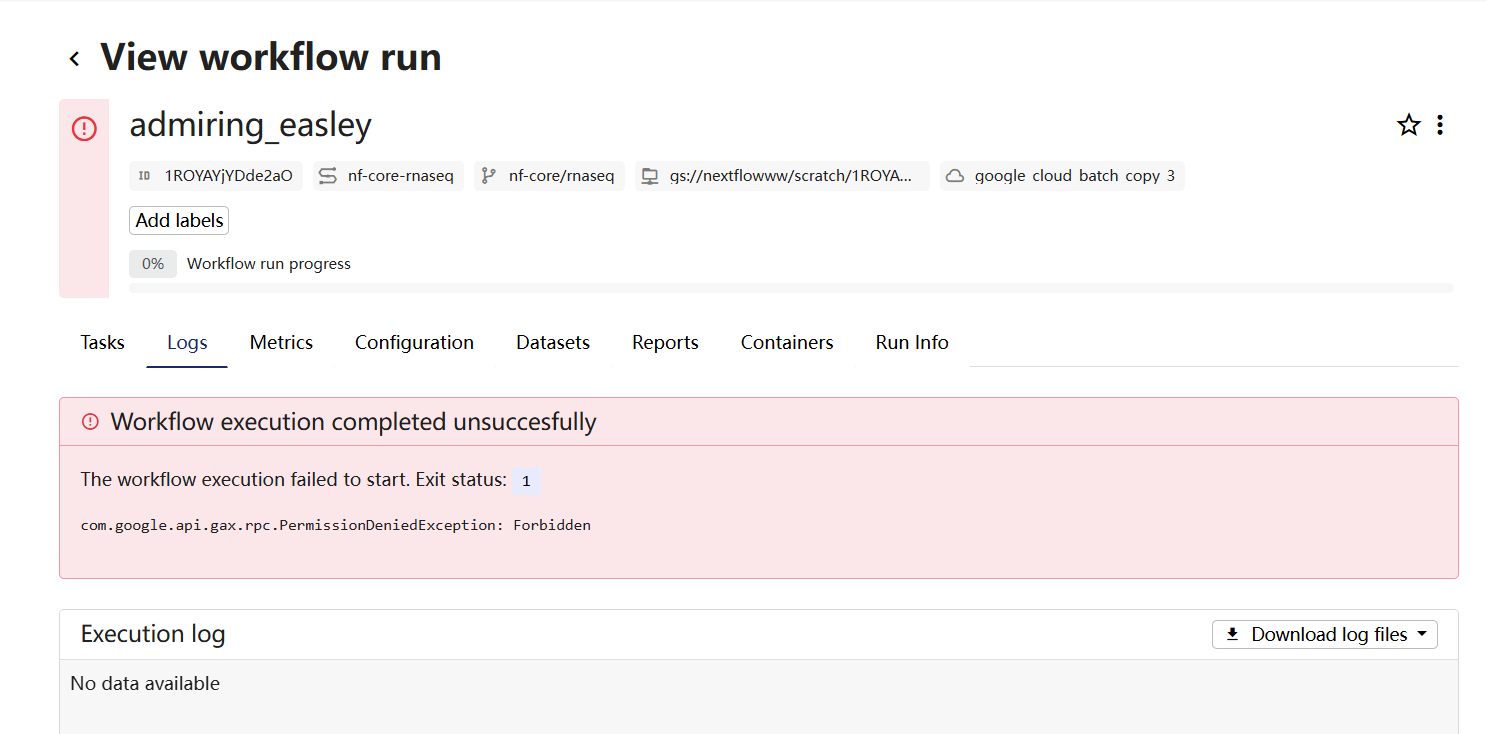
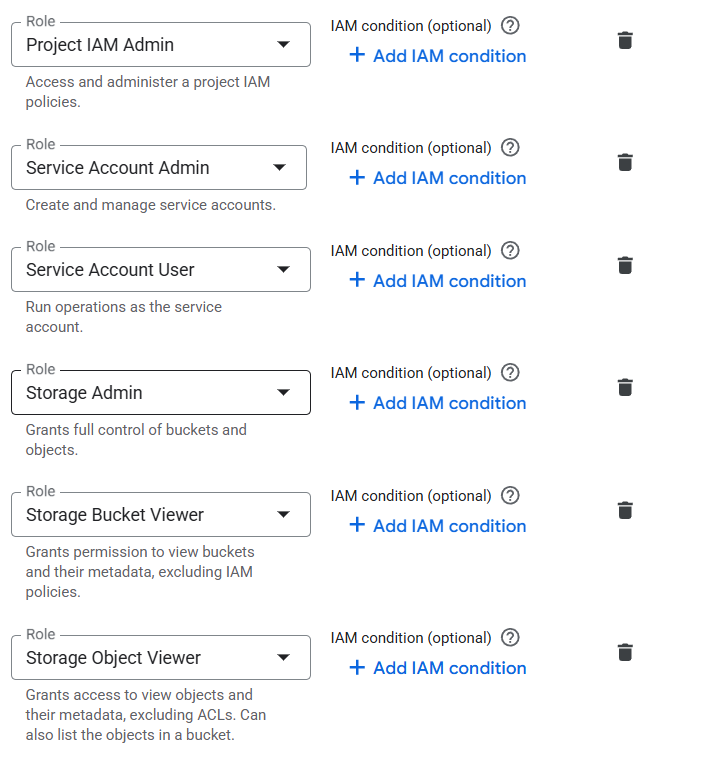
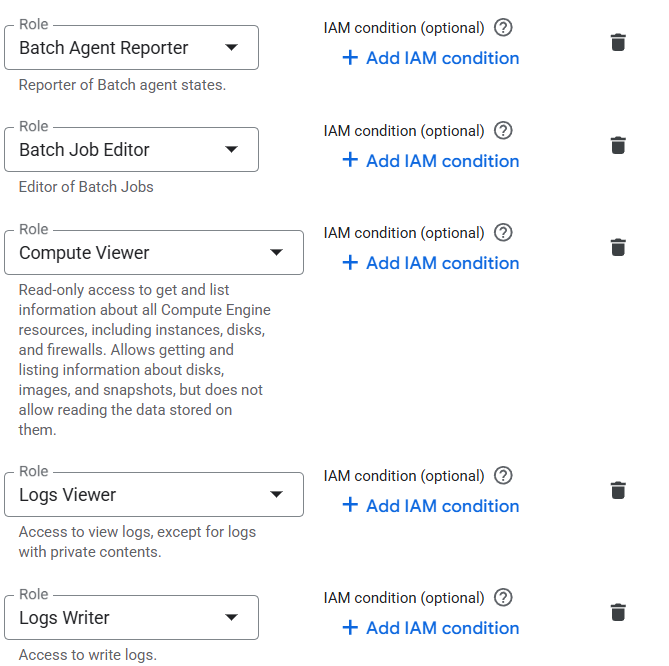
I am new to this platform, just set the environment using Google cloud, I am trying to use my data to run the nf-core-rnaseq pipeline. This keeps jumping out when I use it. I don’t know where I should look for it. I used Seqera AI to check the permissions and granted all permissions as instructed.
I don’t have a lot of computer background, and I would greatly appreciate it if you could help me with it.
I set more admir role on the account, and it seems the problem is solved.
However, if I only use the genome EB2 as a reference, how do I avoid uploading the FASTA genome file and the GTF file? If not, where can I find a GTF file for B. subtilis 168?
here is the log file, many thanks if you can help me with it.